
There goes a lot into the process of making a drug. Before a drug is deemed suitable for patients, it has to go through rigorous testing and cost-effectiveness analyses. In this article, I have tried to explain the whole process, right from the Research and Development to Marketing. This article will be sub-divided into two parts. In this part, I have explained the Research and Development part, and the generally used terminologies in the Pharma Industry. Since this is a bit technical, I have tried to explain everything in layman terms and tried my best not to go off-track.
Initiation
A Drug study generally starts with an analysis of how the disease works and multiplies, which leads to the identification of specific targets. In the later stages, teams of chemists, pharmacologists and biologists engage in screening thousands of compounds or chemically or genetically engineering new ones to generate potential compounds. The molecules that have desirable properties are further modified to enhance the activity or minimize side effects. (this process is known as Lead Optimization)
Testing
After a compound is synthesized, it is being put into trial, Pre Clinical testing (on animals) and Clinical Testing (on Humans). Clinical testing has three phases; the first phase includes a small number of volunteers (20 to 100 people) to determine the safety profile of the drug, the next phase includes a higher number of Volunteers (100-500) to determine the efficacy of the drug. And the last phase of the trials has more than 1000 volunteers to confirm the drug’s effectiveness and if there are any side effects.
After the phase 3 trials, the company submits its research with the Food and Drug Administration (FDA) that is the regulator for approval of the drug, the Application that a company file for approval of a drug is known as a New Drug Application (NDA). NDA is a comprehensive document with 15 sections that explain the whole clinical study, the drugs pharmacology, toxicology and dosage. After getting approval it manufactures and markets the drug.
Patent
More than a Billion Dollars are invested in this whole process and the company wouldn’t want anyone to copy their research and develop the drug themselves. Thus, they apply for a patent which gives them exclusivity to sell in the market. The company generally receives a patent for 20 years and is generally filed during the clinical trials. As the whole ordeal from development to the approval of drugs takes 12-13 years, the company just has 8-10 years left wherein it gets exclusivity to sell the drug.

From Intermediates to Formulation
For manufacturing a good, we need raw materials of a certain kind. In the pharmaceutical industry, it is termed as intermediates. These are chemical compounds, which have to go through various chemical reactions to form a single component, which is known as Active Pharmaceutical Ingredient (API). These are the basic drugs itself which have desired medicinal properties and are also known as bulk drugs. APIs are then converted into a form which can be consumed as a drug by humans known as Formulations; it can be in a form of tablet, syrup, injectables, capsule, patches. A dosage form of a drug is usually composed of two things- API and an excipient (which is the substance of the tablet or the liquid the API is suspended into.)
There are manufacturers who specifically manufacture API or Bulk Drugs and sell them to other companies which then manufacture the formulation and market it. The API manufacturer needs to file a document known as Drug Master File (DMF) with regulatory bodies. The DMF includes information regarding the facilities, processes and intermediates used in the manufacturing of the API.

Generics v/s Patented
When a drug is patented by a company, it develops a brand for that drug in the market and is also known as Branded drugs. Gilead Science, a pharmaceutical company based out of the US has a patent on a drug named Truvada which treats HIV diseases. No other company can sell it in the United States, Whereas in India it does not have a patent and is sold under the name Tenvir-EM by Cipla as it is Generic in India. The drug costs more than $1800 in the US and in India, it costs about $35. Gilead Science generates sales of $3 Billion globally from Truvada and more than 50 % of it comes from the US. If a drug generates sales which are more than a billion dollars it is usually termed as blockbuster drug.
When the drug goes off-patent, it becomes a generic drug from a branded drug and can be sold by any company which gets approval from the FDA of that particular country. If a company sells the drug under the formula name it is termed as unbranded generics whereas if the company sells it under a brand name like Tenvir-EM it is known as Branded generics.

We know that when a company develops a drug, it patents it, and after FDA approval it can sell it in the market. What if another player wants to sell it in the market where the drug is or was patented?
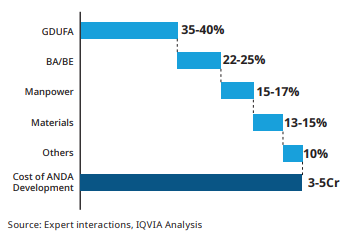
It needs to file an application known as Abbreviated New Drug Application (ANDA) wherein it needs to prove the bioequivalence of the drug. The company is not required to conduct any clinical trials and requires less information than an NDA. An ANDA includes all the information regarding the efficacy, usage and the side effects of the drug. If approved, the company’s drug will be listed in the orange book which includes all the drugs which the FDA has found to be safe and effective. There are four ways through which a company can file for an ANDA
Para 1 Filing – The filing is made during the launch of a generic drug when the innovator has not provided the required info with the orange book.
Para 2 Filing – The application is submitted by the company under Para 2 when the patent is already expired
Para 3 Filing – Para 3 filing is made when the applicant seeks approval from the FDA and will not sell the drug until the patent gets expired
Para 4 Filling – The company files an application under Para 4 when the company thinks the patent is invalid or it will not be infringed by the drug, for which approval is being sought. If the company proves above the two scenarios and gets an approval for the FDA, then it can manufacture and market the drug. The company will receive a 180 days exclusivity wherein even the patented drugs are not sold in the market. The company which has developed the drug and patented it can sue the other company and it can turn into a costly affair for the Generic manufacturer.
Large molecule Drugs vs Small Molecule Drugs
Small molecule drugs (drugs manufactured through chemical synthesis) make the majority of global pharmaceutical sales. There is another form of drugs known as large molecule drugs or biologics, which are protein-based molecules produced by DNA Technology. A medicine derived from a variety of living organisms such as humans, plants, animals, and microorganisms is known as biologics. While most generic drugs are chemically synthesized small molecules, biologic drugs are much more complex in their composition. With the development of biologics comes biosimilars.
Biosimilar is a biologic drug that is quite similar to the FDA approved reference drug. The manufacturer of a proposed biosimilar product generates an array of data comparing the proposed product to the FDA approved reference product in order to demonstrate bio-similarity. The comparative data is generated and evaluated in a step-wise fashion that begins with the foundation of detailed analytical (structural and functional) characterization and comparison of the products, moving on to animal studies, and then to comparative clinical studies.
A generic drug generally has a simplified manufacturing facility compared to biosimilars or biologics. Biosimilars are highly challenging for its manufacturers as the proteins are highly sensitive to change in temperature and humidity condition and so any mild deviation in the planned procedure might lead to degradation or an undesired change to the protein itself. Even though the manufacturing process of biosimilars is rigid, it has been gaining traction due to its promising efficacy when used to treat cancer and autoimmune diseases. Like for small molecule drugs we had an orange book, which lists all the drugs which are approved by the FDA, large molecule drugs have the Purple book.
In-Licensing and Out-Licensing
Licensing is the transfer of rights to a third party to use the intellectual property owned by an innovator. The intellectual property offered by innovators can be of the patent covering the product or process or the right to use the trademark or brand name. In the year 2014, Sun Pharmaceuticals in-licensed a drug from Merck (US Based Pharma Giant) ILumya, a drug used to treat Psoriasis, for $80 million. Wherein Merck had already conducted Phase 1 and Phase 2 trials, in May 2016 Sun Pharma announced positive results for phase 3 trials. Sun pharma spent a total of $320 million on the R&D and in-licensing of the drug and in 2018 Sun pharma got the US FDA approval to market the drug. Under the arrangement between Sun Pharma and Merck, Sun Pharma will have to pay Milestones and Royalties to Merck. Sun Pharma in the year 2019 out-licensed this drug to China Medical Systems (CMS) for the Greater China Market.
Different Therapeutic Segments
There are many doctors who specialise in anatomy, functions and disorders related to specific segments of the human body. There is a neurologist who specialises in disease related to the nervous system, then there is an oncologist who specialises in diseases which are related to different forms of cancer and tumours. Similarly, there are different therapeutic areas or segments like oncology, dermatology, cardiology which a company undertakes and researches to produce different drugs to treat disease-related to specific therapeutic segments.
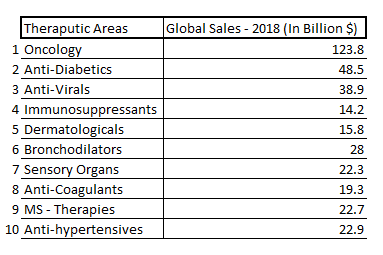
Oncology and Anti-Diabetics have exposure to more than 10% of the global pharmaceutical sales and diseases under these therapeutic areas are one of the major causes of deaths across the world. Chronic prescriptions account for more than two-thirds of prescriptions in the US. Chronic therapies include complex therapies and higher patient out of the pocket expenditure. There has been a move to complex therapies like gene therapy, hormone replacement therapy used to treat different chronic conditions.
Stay tuned for the Second part in which I will explain more about the Indian Pharmaceutical Sector.
Update, 5th June 2020 : How does Pharma Work – Part 2 is now Live.
Subscribe to our Newsletters to get exciting content delivered to your Mailbox!




{kind=link}
Excellent article Gaurav. Kudos to you for such an informative article.
I just have a small question
Do vaccines also follow the same process?
Thank you Rahul!
Yes vaccines also follow the same process. Every innovator drug has to go through pre-clinical and clinical testing to get an approval from the regulator.
Hey Gaurav Great article, love the way you have explained the jargons. I had a question regarding one of the examples u gave about Truvada and Tenvir. Lets say I am a Pharma company in US, and I know about the patent on Truvada, will that stop me from selling the generic of that drug or also manufacturing it, can i mfg the same drug and export it to another country? Also, if i am plainly involved in trading of the drugs, assuming i have all the the required licences, can i import from India the same drug by cipla… Read more »
Thanks Rishab. Generally, the cost of manufacturing in the US is quite more than the cost of manufacturing in emerging markets like India. Indian Pharma Companies have been a Major Player in the Generics Segment. So if you are a US Based Pharma Company manufacturing a patented drug to export it to another country like India, you wouldn’t be able to compete with the domestic manufacturers. The other factor that plays out is to get approval from the FDA of the country in which you want to market the drug. There is altogether a complete article I have written to… Read more »